



Epidemiology
- Specific, hereditary form of bronchiectasis
- Most common genetic disease in the US
- 1:3000 births in Caucasians
- Autosomal recessive with variable penetrance
- CF gene on the long arm of Chromosome 7
- Encodes the CF transmembrane regulator protein (CFTR)
- CFTR located at the cell surface of epithelial cells
- Ion channel that regulates liquid volume on epithelial surfaces
- Affects chloride secretion and inhibition of sodium absorption
- May regulate other cell proteins
- Over 1700 mutations identified
- F508 most common mutation (Class II) -- accounts for 90% of cases of European descent
- deletion of single phenylalanine at position 508
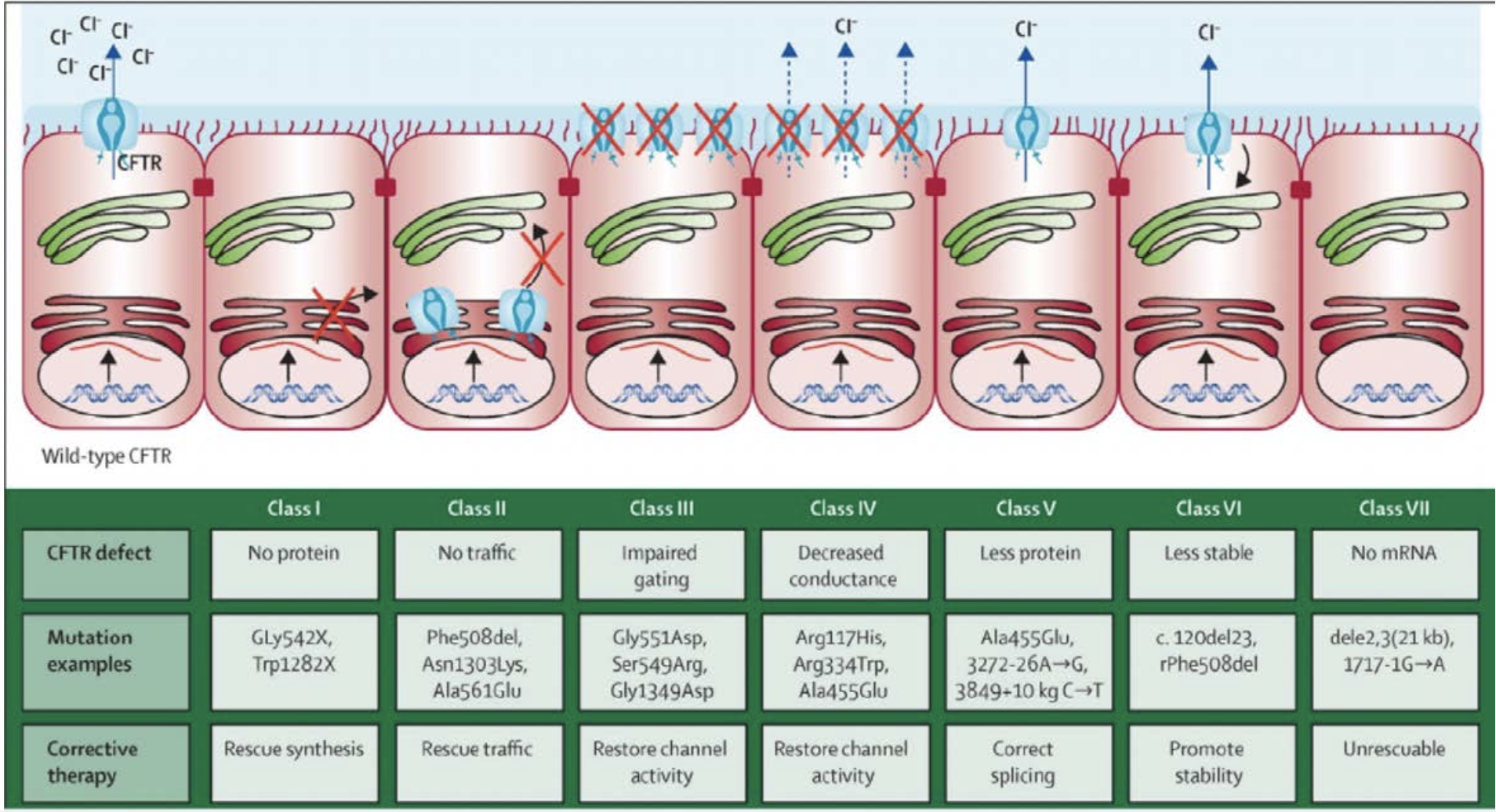
- Class I is most severe
Pathophysiology
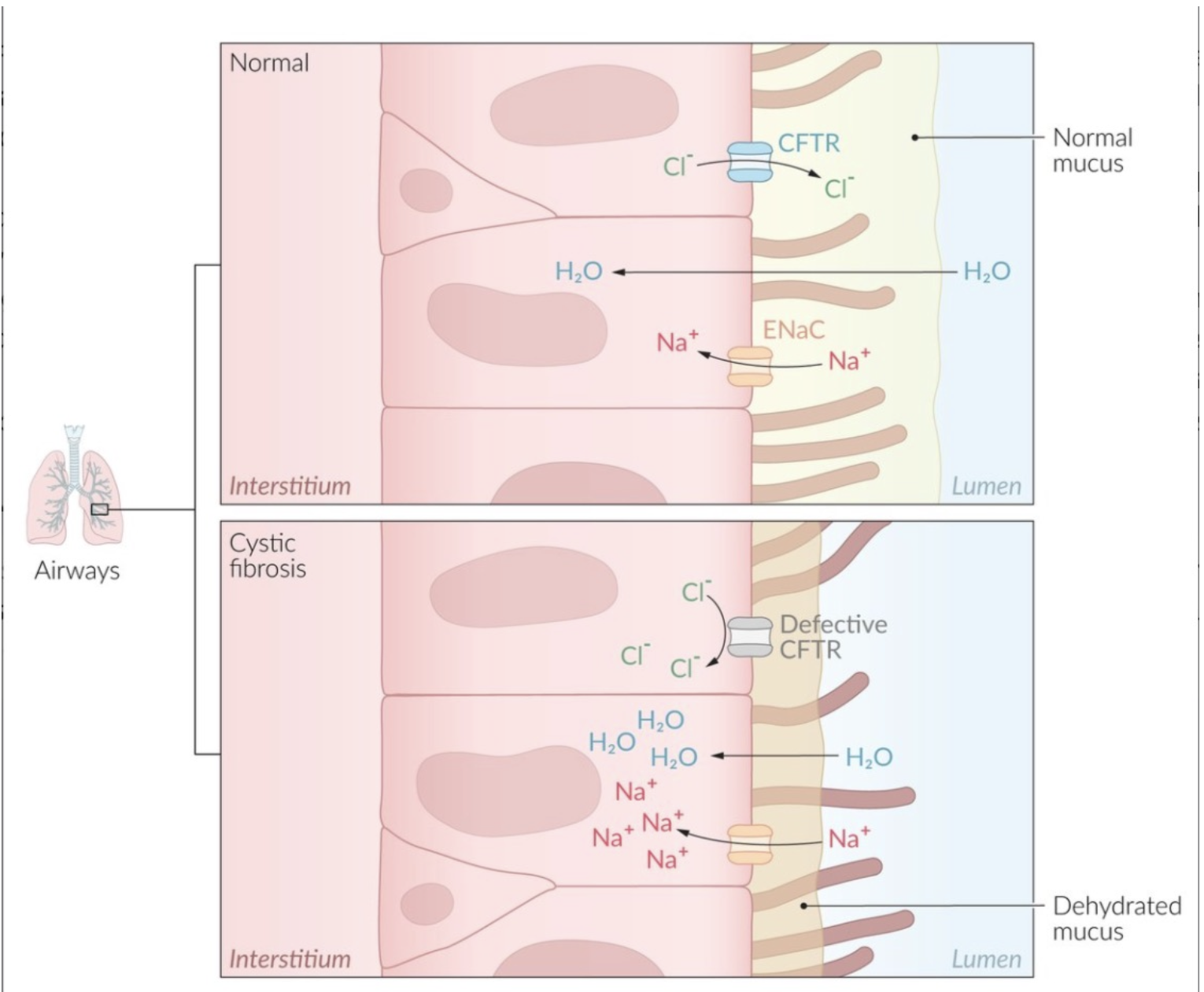
- Also reduced bicarbonate secretion
- Increased viscosity due to impaired mucociliary clearance → persistent bacterial infection → increased inflammation (cellular debri including DNA and elastase) → Airway obstruction → progressive lung dysfunction
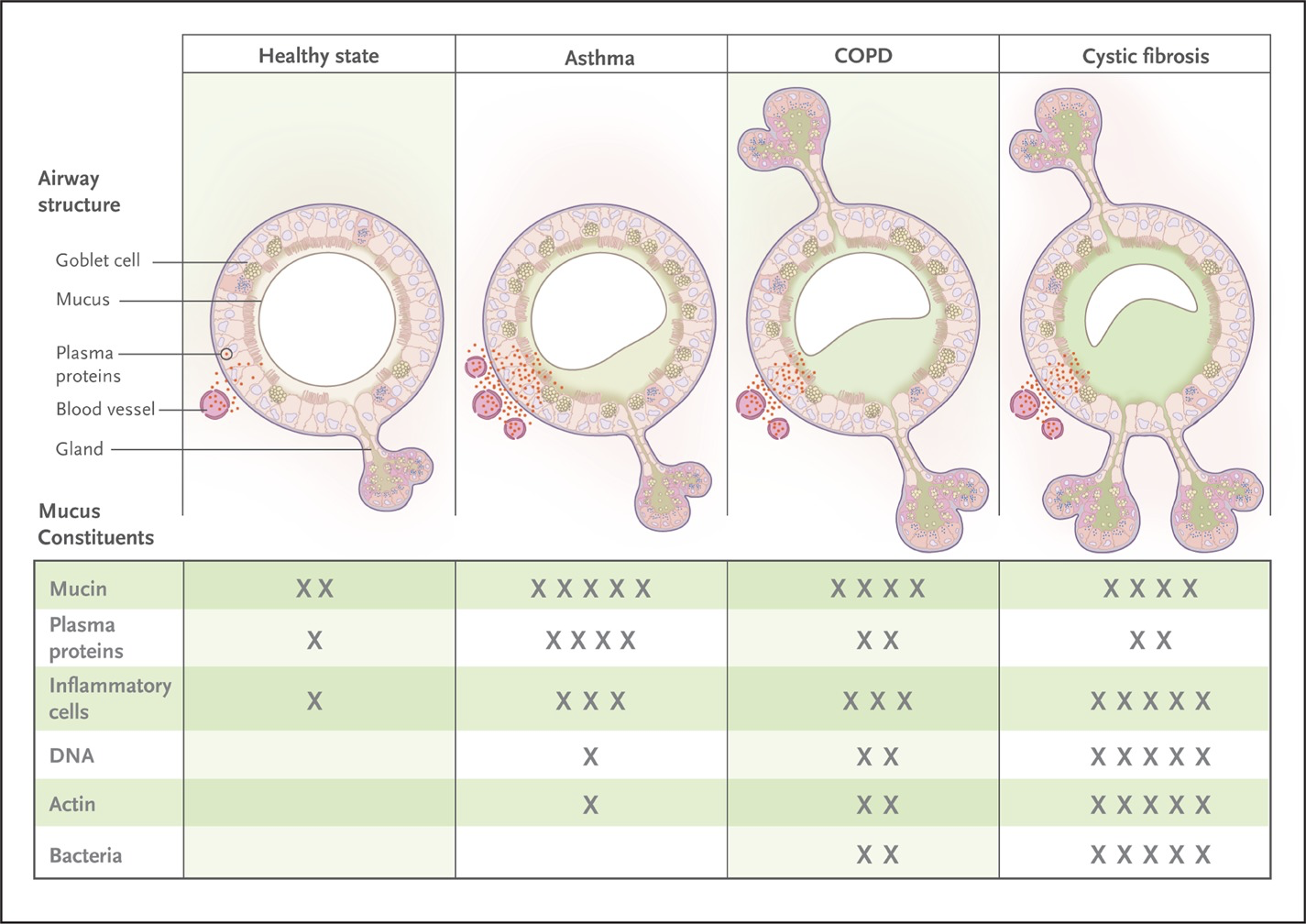
- Fahy JV, Dickey BF. NEJM 2010; 363:2233-47
Diagnosis
- Newborn screening
- Areas where CF is common
- Detects pancreatic enzyme IRT (Immunoreactive Trypsinogen)
- Released into blood when pancreas is damaged (pancreatic damage in utero)
- High levels of IRT = CF confirmed
- Sweat test
- Detects high chloride
- The CFTR enzyme works opposite in lungs & pancreas vs Skin/Sweat Ducts
- In the lungs and pancreas, the chloride cannot exit the cells
- In the skin/sweat ducts, the chloride cannot enter the cells (hence higher levels of chloride in sweat test)
- Diagnostic Criteria
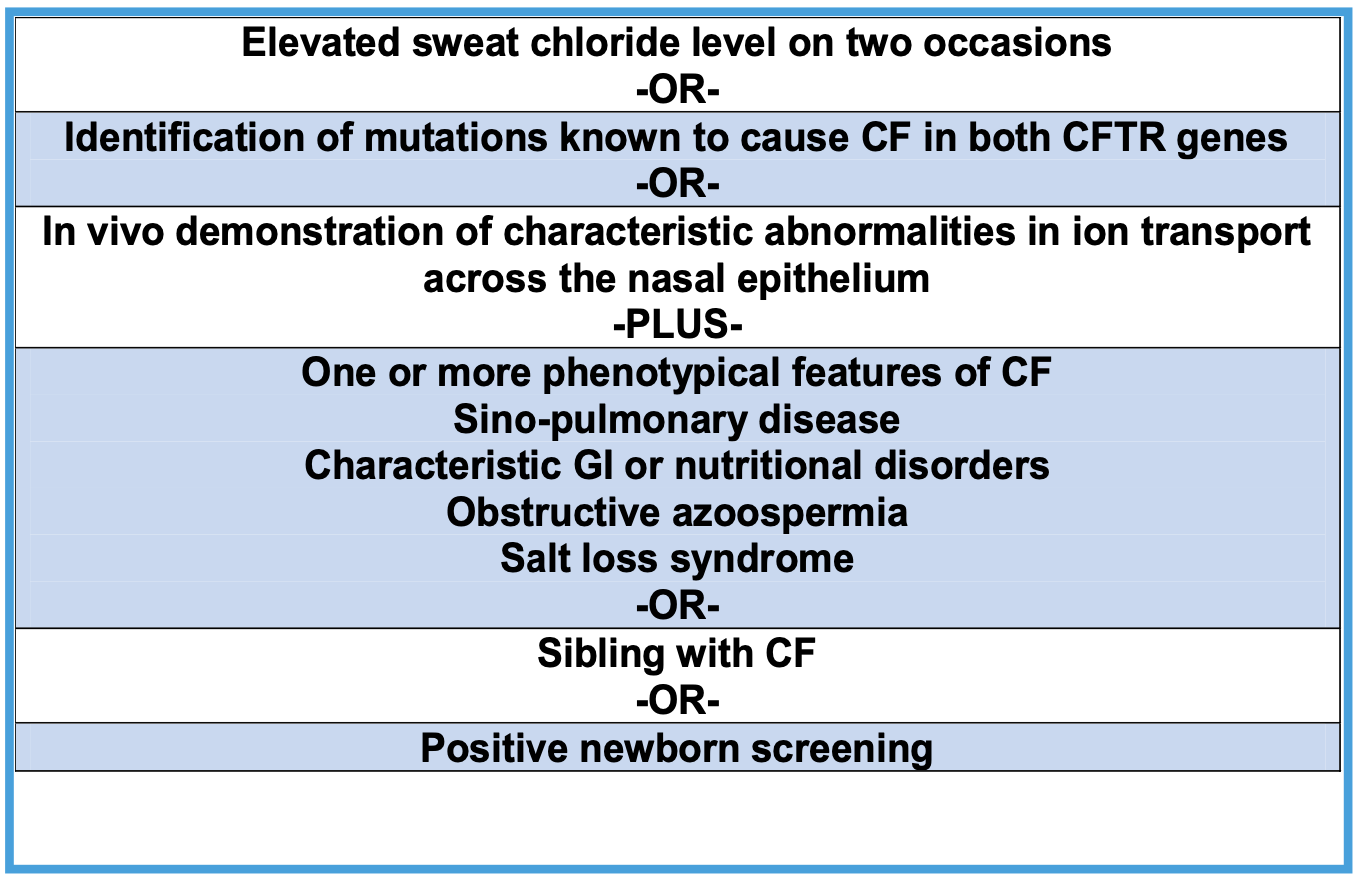
- Per CF Foundation 2015 guidelines
- Normal threshold for sweat chloride change to <30 mmol/L for all ages
- CF-related metabolic syndrome (CFRM) and CF screened positive inconclusive diagnosis (CFSPID) as same entity
- Recommend: repeat sweat chloride testing and close follow up recommended
- The term CFTR related disorder preferred over "atypical" or "non-classical CF" (patients with 0-1 mutations and clinical signs suggestive of CF)
- Usually occurs in adult-onset CF
- Approximately 10% of CF
- CF in the Adult
- Often results from CFTR mutations with residual function, resulting in delayed onset and lesser disease activity
- When to suspect:
- Chronic infection with pseudomonas, S. aureus, NTM
- recurrent/chronic idiopathic pancreatitis
- Bilateral absence of vas deferens
- Nasal polyposis/chronic sinusitis
- Unexplained bronchiectasis
- Criteria
- Presence of CF symptoms AND 1 of the following
- Sweat chloride >60 mmol/L
- Two identified CF-causing mutations
- Sweat chloride 40-59 mmol/L with 0 or 1 CF causing mutation and strong clinical presentation or family history
- Presence of CF symptoms AND 1 of the following
- Diagnosis of CF is very unlikely with sweat chloride <30 mmol/L
Pulmonary Disease in Cystic Fibrosis
- Primary cause of morbidity and mortality in patients with CF is bronchiectasis that leads to obstructive lung disease that ultimately progresses to respiratory failure, pulmonary hypertension and death
- It's present in 98% of patients with CF by the time they reach adulthood
- Death is due to respiratory failure/complications
- Initially may be just a recurrent cough → becomes persistent
- Early on can also see airway hyperreactivity and wheezing but tends to disappear and bronchodilator response tends to disappear as they get older likely due to destruction of lung and cartilage
- Infection
- Early on -- H influenza and S. aureus
- As patient ages -- S. aureus and Pseudomonas
Management
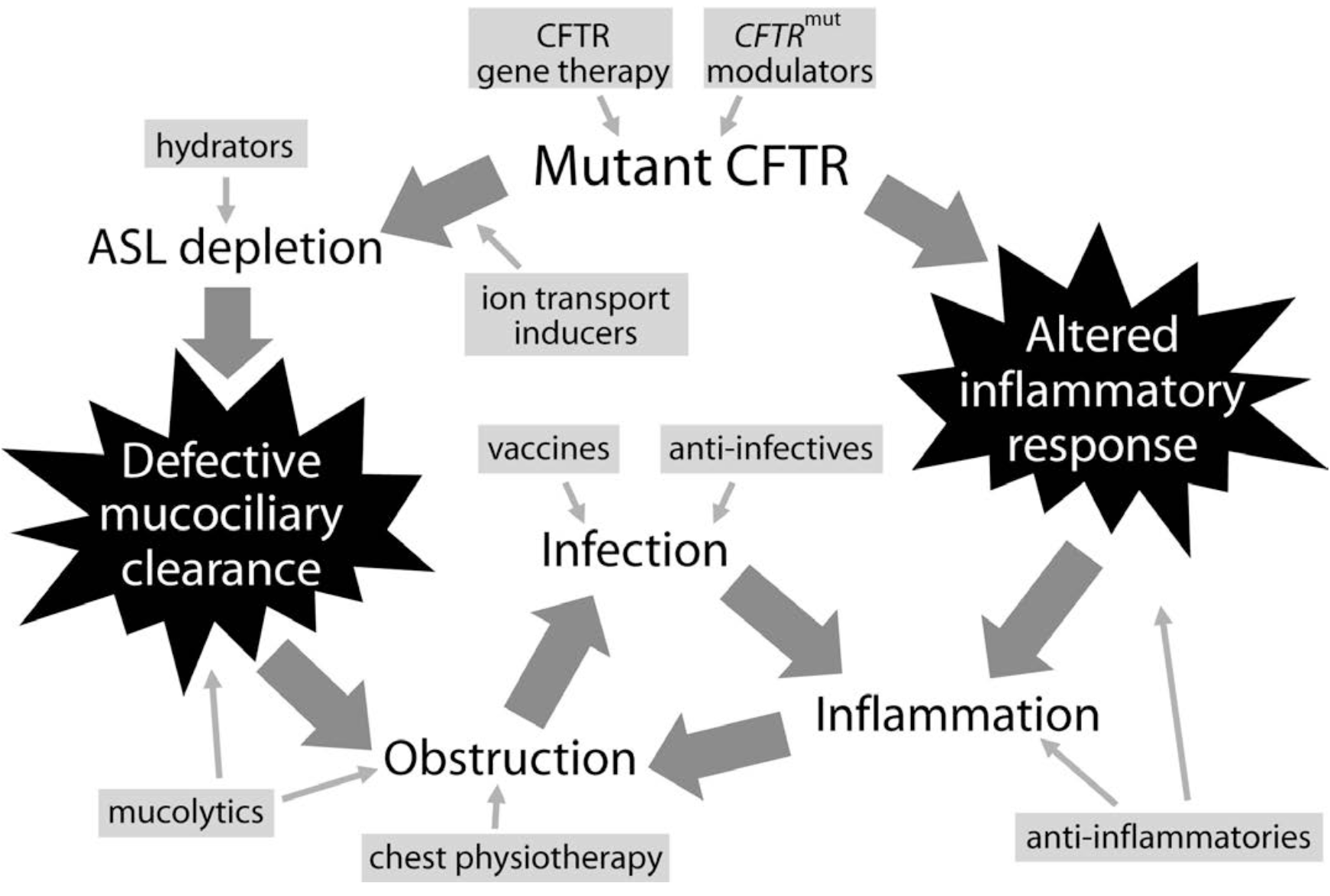
- Flume PA. BMC Medicine 2012; 10:88
- The inflammation seems to be intrinsic and independent from the infection
- CF Modulators
- Enhance the function of the CFTR
- Three modalities: Potentiators, Correctors, Production Correctors
- Potentiators -- increase the funciton of CFTR channels on the cell surface
- Correctors -- improve the processing and delivery of functional CFTR protein to the cell surface.
- This increases the amount of CFTR protein at the cell surface, resulting in enhanced ion transport
- Production Correctors
- or read-through agents promote the read-through of premature termination codons in CFTR mRNA
- Shown to improve pulmonary function, QOL, exacerbations and morbidity and mortality
- Triple Therapy = elexacaftor-tezacaftor-ivacaftor/ETI
- Patients ≥ 12 years F508del homozygote
- Patients ≥ 12 years F508del heterozygote regardless of what is present on their second CFTR allele
- Dual Therapy
- tezacaftor-ivacaftor
- Ages 6-11 F508del homozygote or F508del heterozygote with residual function mutation
- lumacaftor-ivacaftor
- Ages 2-5 F508del homozygote
- tezacaftor-ivacaftor
- Monotherapy
- ivacaftor
- Specifically designed for G551D mutation (gating mutation)
- Impairs the regulated opening of the ion channel
- Patients with at least 1 copy of a gating or residual function mutation not eligible for triple or dual therapy
- Can be used in patients as young as 6 months
- Specifically designed for G551D mutation (gating mutation)
- ivacaftor
- Antibiotic Therapy -- Four approaches/roles
- Chronic prophylaxis
- prevent specific infection
- Not recommended for S aureus
- Conversion to culture negativity upon detection of new specific pathogens (especially Pseudomonas, ? MRSA)
- Eradicate Pseudomonas on detection
- inhaled tobramycin (1 month) or aztreonam strongly recommended for those with moderate to severe reduction in lung function
- Eradicate Pseudomonas on detection
- Palliation of acutely elevated signs and symptoms of infection
- Chronic suppression of established infections
- Not recommended for S aureus
- Established gram negatives
- inhaled tobramycin or aztreonam strongly recommended for those with moderate to severe reduction in lung function
- Insufficient evidence to recommend other inhaled antibiotics (gentamicin, colistin, fluoroquinolones, cephalosporins)
- Chronic prophylaxis
- Bronchodilator Therapy
- Not recommended routinely because most patients lose their responsiveness as they get older
- At least 10% improvement in FEV₁
- Concomitant Asthma or ABPA
- Prior to CPT and inhalational therapy
- ? improve mucociliary clearance (salmeterol may restore chloride secretion)
- Not recommended routinely because most patients lose their responsiveness as they get older
- Anti-Inflammatory Therapy
- Azithromycin thrice weekly recommended in patients over 6 years of age colonized with Pseudomonas
- Considered in >6 years without Pseudomonas
- High dose ibuprofen recommended ages 6-17
- Leukotriene modifiers NOT recommended
- Systemic steroids NOT recommended
- Inhaled corticosteroids for concomitant ABPA or asthma
- Azithromycin thrice weekly recommended in patients over 6 years of age colonized with Pseudomonas
- Non-pharmacologic Therapy
- Clearance of airway secretions recommended for all patients with cystic fibrosis for clearance of sputum, maintenance of lung function, and improved quality of life
- Chest physiotherapy
- forced expiratory techniques
- mechanical vests
- flutter valves
- None proven superior to others
- Aerobic exercise recommended as an adjunct
- Reduce viscosity of secretions
- rhDNase I (dornase α)
- All patients > 6 years
- Inhaled hypertonic saline
- All patients > 6 years
- Inhaled mannitol recommended as second line agent for patients who fail the above because it's irritating to the airways
- rhDNase I (dornase α)
- Non-infectious Pulmonary Complications
- Hemoptysis
- Pneumothorax
- Non-TB
- ABPA
- Respiratory failure; cor pulmonale
- Transplantation in CF
- Generally good candidates
- Transplanted lung does not develop defect
- Requires bilateral lung transplant
- CF Foundation published updated referral guidelines in 2019
- Contraindications follow those in place for lung transplantation in general
- Disease specific contraindications are all relative and vary by center
- Referral
- Patents < 18 years
- FEV₁ <50% predicted and rapidly declining (e.g. >20% relative decline FEV₁ within 12 months)
- FEV₁ <50% predicted with any marker of shortened survival or malnutrition (BMI <10th percentile, while working to improve nutritional status)
- FEV₁ <40% predicted
- Patients ≥ 18 years, alters FEV₁ threshold
- Any of the following regardless of FEV₁
- 6MWT < 400 meters
- Hypoxemia (SpO₂ <88% or PaO₂ <55 mm Hg, at rest or exertion)
- Hypercarbia (PaCO₂ >50 mm Hg, confirmed on ABG)
- Pulmonary artery systolic pressure >50 mm Hg
- Any exacerbation requiring positive pressure ventilation
- Patents < 18 years
- Median survival has gone up; patients living longer